- Structural Variability of Antibody Variable Domains:
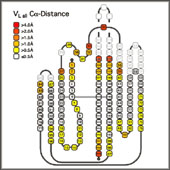
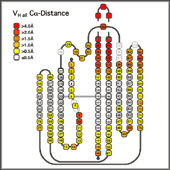
- The x-ray structures of the individual domains were structurally aligned by a least squares fit between the least variable Ca positions (3-7, 20-24, 41-47, 51-57, 78-82, 89-93, 102-108 and 138-144) using the program Insight II (MSI/Biosym). The Ca coordinates of the aligned domains were exported to Excel98 (Microsoft), preserving the new relative orientations. This allowed to the calculation of the mean Ca coordinates for each position and the deviation of the corresponding residue in the actual structures from this consensus position, taking into account the alignments implied by the different numbering schemes. To minimize the effects of the choice of reference structure on the quality of the final alignment, the structural alignment was performed in two steps: first an arbitrary structure was used as a reference structure. From the alignment to this structure, the mean Ca coordinates for each position were calculated and all structures compared to this mean structure. The structure with the lowest deviation from the average was selected as new reference structure and the analysis repeated. The numerical values for the structural deviations were translated into a color code projected onto the sequence alignment
The average deviation can be plotted as a histogram against the residue number . This representation also provides an indication of which positions can truly be considered structurally equivalent (deviation from average Ca position << distance between two neighbouring Ca positions).
|
- Such a coloring of an alignment of individual structures within a family serves well to quickly identify outliers. Larger deviations from the consensus of individual sequences or groups of sequences help to identify residues which cause a conformational change. For instance, a strong structural deviation in the "outer" loop of Vk (L83-L87) from the average Vk conformation correlates with the presence of a non-Gly residue in position L82. This residue usually assumes a positive f torsion angle disallowed for residues with bulkier side chains, and a non-Gly residue in this position results in an outward kink of the loop. The mean Ca deviation for each position, indicated in the header of an alignment of related sequences, serves as a quick reminder of which parts of the sequence represent structurally conserved residues, and which positions are more variable
For example, the variability in positions H9 and H10 of the VH domain can be resolved into four structural subtypes of VH domains, which are determined by the nature of residues H6, H7 and H10. If the average deviation is calculated for each subtype individually, the variability of this region disappears. A similar effect is seen if the VL domains are separated into Vl and Vk domains
|