|
Inhaltsübersicht | Nanomaschinen | Moleküle | Programme | Kurse | Fun | Links |
|
|
| > |
Die Jmol Applikation
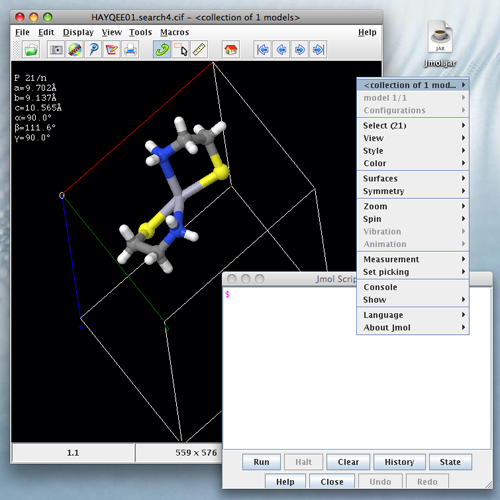
Um auf ihrem Computer lokal beliebige Moleküle mit Jmol abzubilden, können Sie Jmol auch als lokales Programm (Applikation) nutzen.
Kopieren Sie die Datei Jmol.jar auf ihre Festplatte und doppelklicken Sie darauf, um Jmol als eigenständiges Programm zu öffnen.
Der Screenshot links zeigt die Jmol als eigenständiges, lokal installiertes Programm. Neben Kontext-menü und Konsole, die wie im Applet aufgerufen werden, besitzt die Applikation zusätzlich einen Menubalken.
Jmol kann viele verschiedene Typen von Molekül-Koordinatenfiles interpretieren. Die gebräuchlichsten sind das .pdb und .mmcif-Format für makromolekulare Strukturen (PDB Datenbank) und .cif, .mol und .xyz für kleinere Moleküle. Sie haben mehrere Wege, wie sie ein Koordinaten-File mit Jmol öffnen können:
- Drag-and-Drop: Ziehen Sie das File mit gedrückter Maustaste auf das Jmol-Fenster und lassen sie es dort los. Das ist die einfachste Methode.
- Ueber das File-Menü: Unter "File/Open" können Sie Ihre Verzeichnisse nach Ihrem Koordinatenfile durchsuchen.
- Ueber die Konsole: Mit einem Klick auf die rechte Maustaste, <Cntl>-Klick oder Klick auf das jmol-logo in der rechten unteren Ecke des Fensters öffnen Sie ein Kommandokonsolen-Fenster, in das Sie ihre Befehle eintippen können:
"load filepath/filename" öffnet das Koordinatenfile.
Defaultmässig wird eine frisch eingelesene Struktur in der "Stick-and-Ball"-Darstellung angezeigt: Die Bindungen als Zylinder und die Atome als kleine Kugeln, angefärbt nach den verschiedenen Elementen (Kohlenstoff, grau; Sauerstoff, rot; Stickstoff, blau; Wasserstoff, weiss, Schwefel, gelb...). Für Makromoleküle ist diese Darstellungsweise eher unübersichtlich, es gibt jedoch auch abstraktere Darstellungsweisen.
Einige der vielen über die Kommandokonsole verfügbaren Befehle und Darstellungsoptionen sind im nächsten Tutorial beschrieben, eine vollständige Dokumentation finden Sie in der "Jmol interactive scripting documentation" von R. Hanson.