|
Inhaltsübersicht | Nanomaschinen | Moleküle | Programme | Kurse | Fun | Links |
|
|
| > |
Phenylalanine Hydroxylase
The Protein Alphabet
The proteins that make up the skin, muscle, hair, bones and other organs in your body are primarily composed of a set of 20 building blocks, called amino acids. Amino acids are the alphabet in the protein language: when combined in a specific order, they make up meaningful structures (proteins) with varied and specific functions. Amino acids have distinct shapes, sizes, charges and other characteristics. Many amino acids are synthesized in your body from breakdown products of sugars and fats, or are converted from other amino acids by the action of specific enzymes. However, a few of them, called essential amino acids, cannot be synthesized or converted in your body and have to be obtained from the food you eat. Phenylalanine is one such essential amino acid. It is closely related to another amino acid, tyrosine, which just has an additional hydroxyl (OH) group. Liver cells contain an enzyme called phenylalanine hydroxylase, which can add this group and convert phenylalanine to tyrosine. Thus as long as this enzyme is functional and there is a reasonable supply of phenylalanine, tyrosine can be synthesized in your body and does not have to be included in the food that you eat.Phenylalanine Hydroxylase
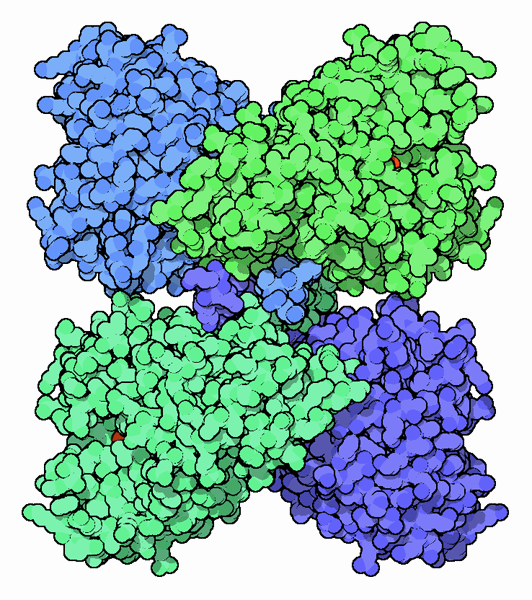
Four molecules of phenylalanine hydroxylase interact to form a tetramer, which is the functional unit for this enzyme. Each molecule in the tetramer is organized into three domains: a regulatory domain, a catalytic domain where the enzyme activity resides and a tetramerization domain that assembles four chains into the tetramer. At the heart of each catalytic domain is an iron ion that plays an important role in the enzyme action. A model structure of the complete enzyme tetramer is shown here. This is composed of two PDB files: PDB entry 2pah, which includes the structure of the catalytic and tetramerization domains of the enzyme, and PDB entry 1phz, which includes the regulatory domain flexibly attached to the catalytic domain.Phenylketonuria
Surprisingly, we usually get too much phenylalanine in our diet, and this can cause problems. Normally, phenylalanine hydroxylase regulates the clearance of about 75% of the excess phenylalanine from our body by converting it to tyrosine. But in 1934, a Norwegian doctor, Asbjorn Folling, showed that the urine of two of his young mentally handicapped patients had a high level of phenylalanine. These were the first diagnosed cases of phenylketonuria. Within the next few years it was shown that the absence or malfunction of phenylalanine hydroxylase leads to this serious genetic disorder. However, it was not until the early 1950s that this information was put to use. A child suffering from phenylketonuria was treated with a diet low in the amino acid phenylalanine, showing that many of the symptoms of this disease could be reversed if treated early. Presently, in many places, newborn children are routinely screened for phenylketonuria and placed on a low phenylalanine diet if diagnosed. As these children grow up, they are also recommended to avoid the sweetener Aspartame, since it contains phenylalanine. In a few cases, lack of the tetrahydrobiopterin, an essential cofactor for the enzyme, or an inability to regenerate it can also lead to phenylketonuria. These cases may be treated with tetrahydrobiopterin supplements.Next: Aromatic Alliance
Last changed by: A.Honegger,